Structure prediction is a fundamental problem in molecular science because the structure of a molecule determines its properties and functions. In recent years, deep learning methods have made remarkable progress and impact on predicting molecular structures, especially for protein molecules. Deep learning methods, such as AlphaFold and RoseTTAFold, have achieved unprecedented accuracy in predicting the most probable structures for proteins from their amino acid sequences and have been hailed as a game changer in molecular science. However, this method provides only a single snapshot of a protein structure, and structure prediction cannot tell the complete story of how a molecule works.
Proteins are not rigid objects; they are dynamic molecules that can adopt different structures with specific probabilities at equilibrium. Identifying these structures and their probabilities is essential in understanding protein properties and functions, how they interact with other proteins, and the statistical mechanics and thermodynamics of molecular systems. Traditional methods for obtaining these equilibrium distributions, such as molecular dynamics simulations or Monte Carlo sampling (which uses repeated random sampling from a distribution to achieve numerical statistical results), are often computationally expensive and may even become intractable for complex molecules. Therefore, there is a pressing need for novel computational approaches that can accurately and efficiently predict the equilibrium distributions of molecular structures from basic descriptors.
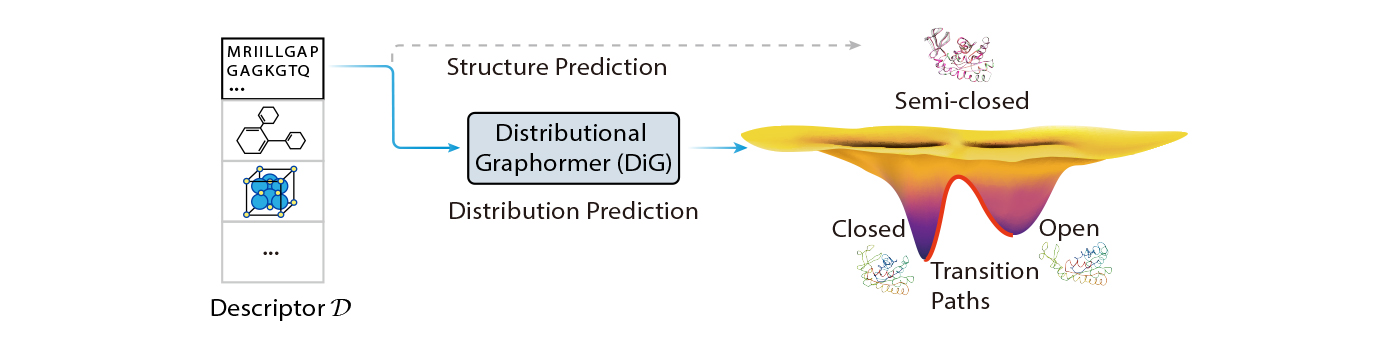
In this blog post, we introduce Distributional Graphormer (DiG), a new deep learning framework for predicting protein structures according to their equilibrium distribution. It aims to address this fundamental challenge and open new opportunities for molecular science. DiG is a significant advancement from single structure prediction to structure ensemble modeling with equilibrium distributions. Its distribution prediction capability bridges the gap between the microscopic structures and the macroscopic properties of molecular systems, which are governed by statistical mechanics and thermodynamics. Nevertheless, this is a tremendous challenge, as it requires modeling complex distributions in high-dimensional space to capture the probabilities of different molecular states.
DiG achieves a novel solution for distribution prediction through an advancement of our previous work, Graphormer, which is a general-purpose graph transformer that can effectively model molecular structures. Graphormer has shown excellent performance in molecular science research, demonstrated by applications in quantum chemistry and molecular dynamics simulations, as reported in our previous blog posts (see here and here for more details). Now, we have advanced Graphormer to create DiG, which has a new and powerful capability: using deep neural networks to directly predict target distribution from basic descriptors of molecules.
SPOTLIGHT: AI focus area

AI and Microsoft Research
Learn more about the breadth of AI research at Microsoft
DiG tackles this challenging problem. It is based on the idea of simulated annealing, a classic method in thermodynamics and optimization, which has also motivated the recent development of diffusion models that achieved remarkable breakthroughs in AI-generated content (AIGC). Simulated annealing produces a complex distribution by gradually refining a simple distribution through the simulation of an annealing process, allowing it to explore and settle in the most probable states. DiG mimics this process in a deep learning framework for molecular systems. AIGC models are often based on the idea of diffusion models, which are inspired by statistical mechanics and thermodynamics.
DiG is also based on the idea of diffusion models, but we bring this idea back to thermodynamics research, creating a closed loop of inspiration and innovation. We imagine scientists someday will be able to use DiG like an AIGC model for drawing, inputting a simple description, such as an amino acid sequence, and then using DiG to quickly generate realistic and diverse protein structures that follow equilibrium distribution. This will greatly enhance scientists’ productivity and creativity, enabling novel discoveries and applications in fields such as drug design, materials science, and catalysis.
How does DiG work?
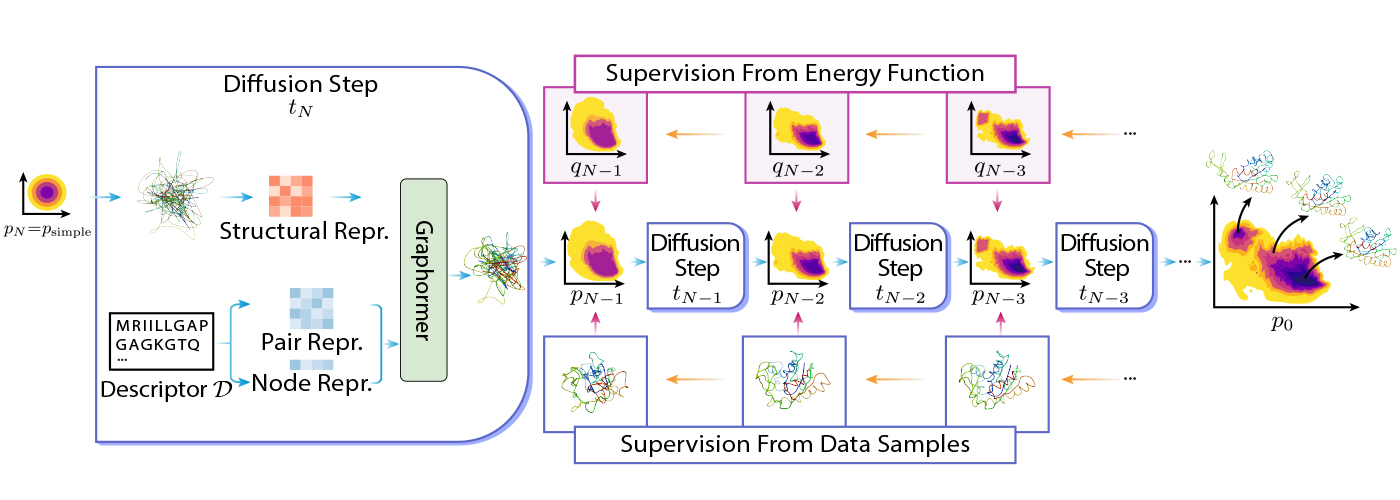
DiG is based on the idea of diffusion by transforming a simple distribution to a complex distribution using Graphormer. The simple distribution can be a standard Gaussian, and the complex distribution can be the equilibrium distribution of molecular structures. The transformation is done step-by-step, where the whole process mimics the simulated annealing process.
DiG can be trained using different types of data or information. For example, DiG can use energy functions of molecular systems to guide transformation, and it can also use simulated structure data, such as molecular dynamics trajectories, to learn the distribution. More concretely, DiG can use energy functions of molecular systems to guide transformation by minimizing the discrepancy between the energy-based probabilities and the probabilities predicted by DiG. This approach can leverage the prior knowledge of the system and train DiG without stringent dependency on data. Alternatively, DiG can also use simulation data, such as molecular dynamics trajectories, to learn the distribution by maximizing the likelihood of the data under the DiG model.
DiG shows similarly good generalizing abilities on many molecular systems compared with deep learning-based structure prediction methods. This is because DiG inherits the advantages of advanced deep-learning architectures like Graphormer and applies them to the new and challenging task of distribution prediction. Once trained, DiG can generate molecular structures by reversing the transformation process, starting from a simple distribution and applying neural networks in reverse order. DiG can also provide the probability estimation for each generated structure by computing the change of probability along the transformation process. DiG is a flexible and general framework that can handle different types of molecular systems and descriptors.
Results
We demonstrate DiG’s performance and potential through several molecular sampling tasks covering a broad range of molecular systems, such as proteins, protein-ligand complexes, and catalyst-adsorbate systems. Our results show that DiG not only generates realistic and diverse molecular structures with high efficiency and low computational costs, but it also provides estimations of state densities, which are crucial for computing macroscopic properties using statistical mechanics. Accordingly, DiG presents a significant advancement in statistically understanding microscopic molecules and predicting their macroscopic properties, creating many exciting research opportunities in molecular science.
One major application of DiG is to sample protein conformations, which are indispensable to understanding their properties and functions. Proteins are dynamic molecules that can adopt diverse structures with different probabilities at equilibrium, and these structures are often related to their biological functions and interactions with other molecules. However, predicting the equilibrium distribution of protein conformations is a long-standing and challenging problem due to the complex and high-dimensional energy landscape that governs probability distribution in the conformation space. In contrast to expensive and inefficient molecular dynamics simulations or Monte Carlo sampling methods, DiG generates diverse and functionally relevant protein structures from amino acid sequences at a high speed and a significantly reduced cost.
DiG can generate multiple conformations from the same protein sequence. The left side of Figure 3 shows DiG-generated structures of the main protease of SARS-CoV-2 virus compared with MD simulations and AlphaFold prediction results. The contours (shown as lines) in the 2D space reveal three clusters sampled by extensive MD simulations. DiG generates highly similar structures in clusters II and III, while structures in cluster I are undersampled. In the right panel, DiG-generated structures are aligned to experimental structures for four proteins, each with two distinguishable conformations corresponding to unique functional states. In the upper left, the Adenylate kinase protein has open and closed states, both well sampled by DiG. Similarly, for the drug transport protein LmrP, DiG also generates structures for both states. Here, note that the closed state is experimentally determined (in the lower-right corner, with PDB ID 6t1z), while the other is the AlphaFold predicted model that is consistent with experimental data. In the case of human B-Raf kinase, the major structural difference is localized in the A-loop region and a nearby helix, which are well captured by DiG. The D-ribose binding protein has two separated domains, which can be packed into two distinct conformations. DiG perfectly generated the straight-up conformation, but it is less accurate in predicting the twisted conformation. Nonetheless, besides the straight-up conformation, DiG generated some conformations that appear to be intermediate states.
Another application of DiG is to sample catalyst-adsorbate systems, which are central to heterogeneous catalysis. Identifying active adsorption sites and stable adsorbate configurations is crucial for understanding and designing catalysts, but it is also quite challenging due to the complex surface-molecular interactions. Traditional methods, such as density functional theory (DFT) calculations and molecular dynamics simulations, are time-consuming and costly, especially for large and complex surfaces. DiG predicts adsorption sites and configurations, as well as their probabilities, from the substrate and adsorbate descriptors. DiG can handle various types of adsorbates, such as single atoms or molecules being adsorbed onto different types of substrates, such as metals or alloys.
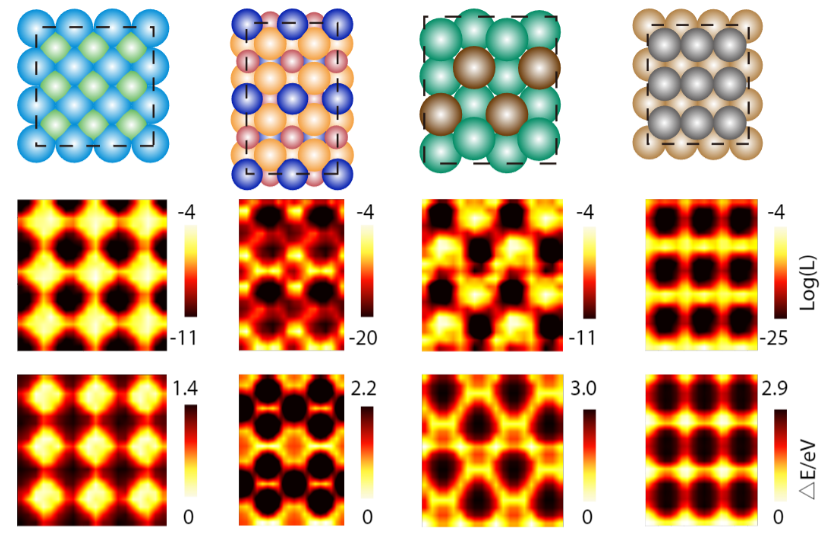
Applying DiG, we predicted the adsorption sites for a variety of catalyst-adsorbate systems and compared these predicted probabilities with energies obtained from DFT calculations. We found that DiG could find all the stable adsorption sites and generate adsorbate configurations that are similar to the DFT results with high efficiency and at a low cost. DiG estimates the probabilities of different adsorption configurations, in good agreement with DFT energies.
Conclusion
In this blog, we introduced DiG, a deep learning framework that aims to predict the distribution of molecular structures. DiG is a significant advancement from single structure prediction toward ensemble modeling with equilibrium distributions, setting a cornerstone for connecting microscopic structures to macroscopic properties under deep learning frameworks.
DiG involves key ML innovations that lead to expressive generative models, which have been shown to have the capacity to sample multimodal distribution within a given class of molecules. We have demonstrated the flexibility of this approach on different classes of molecules (including proteins, etc.), and we have shown that individual structures generated in this way are chemically realistic. Consequently, DiG enables the development of ML systems that can sample equilibrium distributions of molecules given appropriate training data.
However, we acknowledge that considerably more research is needed to obtain efficient and reliable predictions of equilibrium distributions for arbitrary molecules. We hope that DiG inspires additional research and innovation in this direction, and we look forward to more exciting results and impact from DiG and other related methods in the future.
The post Distributional Graphormer: Toward equilibrium distribution prediction for molecular systems appeared first on Microsoft Research.